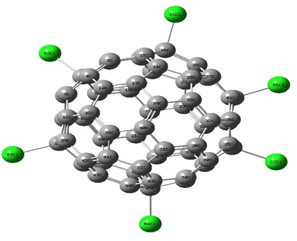
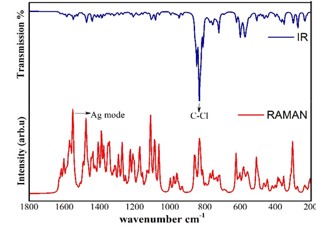
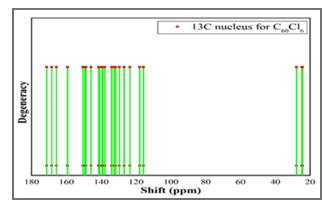
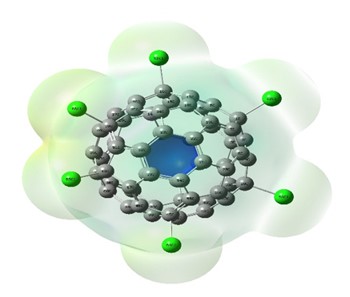
Vibrational Spectroscopic, 13C NMR, DFT Studies on Chlorofullerene (C60Cl6): A Potential Bioactive Agent
Christy P1*, Peter A2
DOI:10.61343/jcm.v1i02.45
1* P Anto Christy, Pg And Research, Department Of Physics, Arts And Science College, Madurai 625019, Tamil Nadu, India.
2 A John Peter, PG and Research, Department of Physics, Arts and Science College, Madurai 625019, Tamil Nadu, India.
The present work comprises the systematic computational chemical findings on Chlorofullerene (C60Cl6). The molecular arrangement of C60Cl6 was augmented by density function theory DFT model B3LYP principle through 6-31G(d,p) basis set using Gaussian 09 program. The infrared and Raman spectra were simulated and assigned for C60Cl6 molecule. Carbon – chlorine stretching vibrations are found to be in the range 150-900 cm-1 and the chains are strongly affected with the radial vibrations of the carbon sphere. The augmented geometries at ground-state of the molecules are calculated without any geometrical restriction and the molecules are found to be minima on their respective potential energy surfaces. The augmented structures have been subjected to Gauge including atomic orbital (GIAO), the chemical shielding tensors applying B3LYP principle through 6-31G(d,p) in solvent phase in order to calculate 13C chemical shift values with respect to trimethylsilane (TMS) as computational reference. Molecular acuteness and constancy were examined using the Frontier molecular orbitals (FMO) examination. The molecular electrostatic potential (MEP) mapping provides a valuable information regarding the net electrostatic effect produced by net charge distribution of the molecule. Chlorofullerenes are considered to be promising compounds for the investigation of biological action which show pronounced anti-HIV action and low toxicity. Hence, these results set goal in scheming the biocompatible molecules which will be beneficial in the field of carbon nano medicine and drug delivery application.
Keywords: DFT, GIAO, Chemical Shift, Electrostatic Potential.
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Pg And Research, Department Of Physics, Arts And Science College, Madurai 625019, Tamil Nadu, India. Email:  |
Christy P, Peter A, Vibrational Spectroscopic, 13C NMR, DFT Studies on Chlorofullerene (C60Cl6): A Potential Bioactive Agent. J.Con.Ma. 2023;1(2):191-194. Available From https://jcm.thecmrs.in/index.php/j/article/view/45 |
 |
 ©
©