Ab Initio Study of Structural and Magnetic Properties of Cobalt Doped Zinc Oxide
Sharmaa Y1, Heera P2*
DOI:10.61343/jcm.v1i02.36
1 Yojana Sharmaa, Department Of Physics Astronomical Science, Central University Of Himachal Pradesh176206, HP, India.
2* Pawan Heera, Department Of Physics Astronomical Science, Central University Of Himachal Pradesh176206, HP, India.
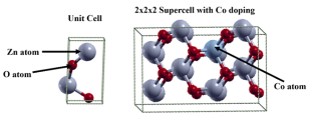
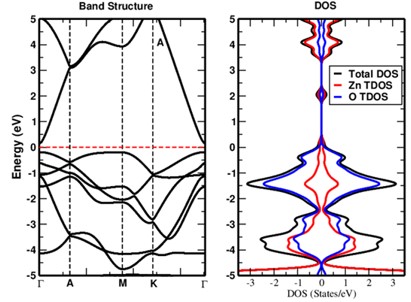
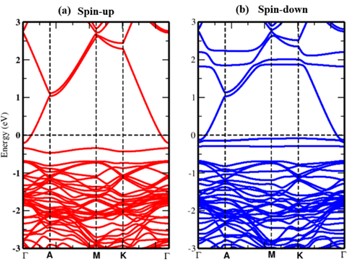
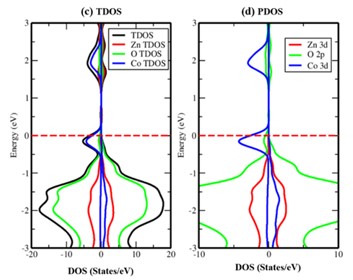
In this study, we have investigated the electronic and magnetic properties of Cobalt doped zinc oxide (ZnO) bulk materials using density functional theory (DFT) calculations. The substitution of Co atoms into the ZnO lattice is systematically explored to understand its impact on the electronic band structure, magnetic moments and stability of the material. It is observed that Co doping introduces localized magnetic moments of 3.073 μB associated with Co atoms. The electronic band structure exhibits the semi-metallic nature after the Co doping with a majority spin (↑) and a minority spin (↓) magnetic configuration hybridization between Co 3d and 2p orbitals of O, indicating the potential for spin-polarized transport in Co-doped ZnO. We have also analyzed the formation energies to assess the thermodynamic stability. It is observed that the binding energy per atom slightly decreased to -3.563 eV, indicating stable Co incorporation. From the obtained results it can be concluded that, through controlled doping strategies the electronic and magnetic properties in ZnO-based materials can be altered for desired properties.
Keywords: Band Structure, Magnetic Moment, DFT (Density Functional Theory), Thermodynamic Stability, ZnO
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , , Department Of Physics Astronomical Science, Central University Of Himachal Pradesh176206, , HP, India. Email:  |
Sharmaa Y, Heera P, Ab Initio Study of Structural and Magnetic Properties of Cobalt Doped Zinc Oxide. J.Con.Ma. 2023;1(2):118-122. Available From https://jcm.thecmrs.in/index.php/j/article/view/36 |
 |
 ©
©