Comparative Analysis of Fold Angles and Aromaticity in Synthetic Models and the Crystal Structure of Nitrate Reductase Enzyme
Periyasamy G1, Lenkennavar S2*
DOI:10.61343/jcm.v1i02.31
1 Ganga Periyasamy, Department Of Chemistry, Bangalore University, Bangalore, Karnataka, India.
2* Susheela K Lenkennavar, Department of Physics, Bangalore University, Bangalore 560 056, India.
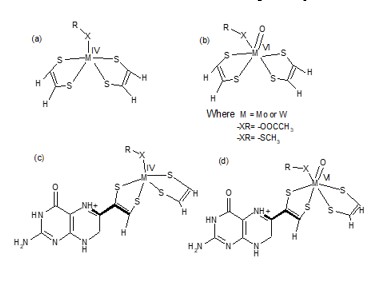
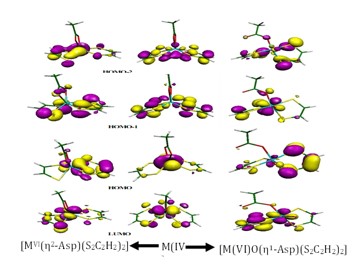
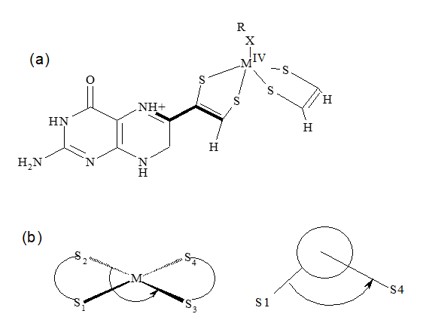
Many research groups aim to synthesize biomimetic compounds to mimic enzymatic functions. To achieve a complete mimicry, a comprehensive understanding of the structural details at the microscopic level is necessary. In this study, we conducted a structural analysis of synthetic molybdenum-dithiolene complexes and the active site structure of the NIR enzyme. Our analysis focused on the folding of the dithiolene ring and the aromaticity of the five-membered ring.
Keywords: NIR enzyme, Biomimetic compounds, dithiolene unit, DFT.
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , , Department of Physics, Bangalore University, Bangalore 560 056, India, . Email:  |
Periyasamy G, Lenkennavar S, Comparative Analysis of Fold Angles and Aromaticity in Synthetic Models and the Crystal Structure of Nitrate Reductase Enzyme. J.Con.Ma. 2023;1(2):142-146. Available From https://jcm.thecmrs.in/index.php/j/article/view/31 |
 |
 ©
©