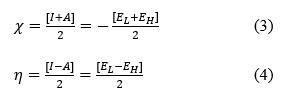Understanding The Behavior Of 5, 10, 15, 20-Tetrakis (4 -Hydroxyphenyl) Porphyrin and Its Cation in Methanol: Insights from Electronic Structure Calculations
Anju1*, Saini L2, Pandey M3
DOI:10.61343/jcm.v1i02.27
1* Anju, Department Of Physics, Sardar Vallabhbhai National Institute Of Technology, Surat 395007, India.
2 LK Saini, Department of Physics, Sardar Vallabhbhai National Institute of Technology, Surat 395007, INDIA.
3 Mukesh Pandey, Atomic and Molecular Physics Division Bhabha Atomic Research Centre, Mumbai 400085, INDIA.
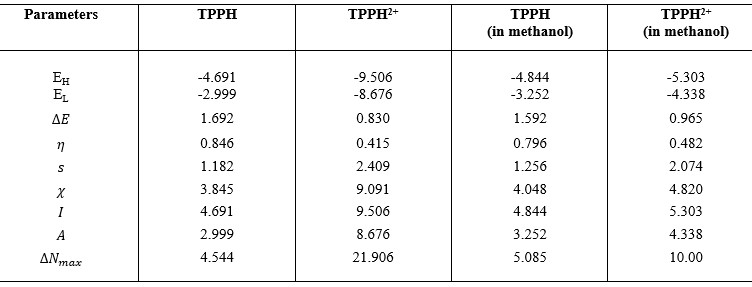
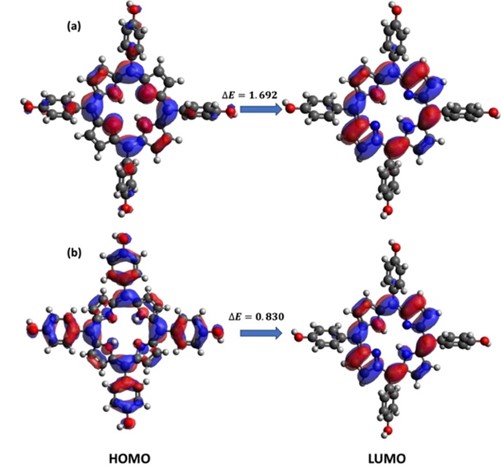
This research investigates the solvation dynamics and interactions of neutral 5,10,15,20-tetrakis(4-hydroxyphenyl) porphyrin (TPPH) and its cationic form (TPPH2+) with methanol as the solvent. HOMO-LUMO analysis and Global Chemical Reactive Descriptors (GCRD) results were reported using DFT method with BP86 functional. The study reveals contrasting charge transfer behaviors: neutral TPPH demonstrates an enhanced charge transfer rate upon dissolution in methanol, while cationic TPPH exhibits a reverse trend. This solvation-induced reduction in energy gap presents a potential avenue for optimizing optoelectronic devices like light-emitting diodes and laser diodes. These findings elucidate the intricate interplay between porphyrin derivatives and solvents, offering valuable insights for tailored applications across diverse scientific and technological fields.
Keywords: Solvation, porphyrin, DFT, energy gap, charge transfer.
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , , Department Of Physics, Sardar Vallabhbhai National Institute Of Technology, Surat 395007, , India. Email:  |
Anju, Saini L, Pandey M, Understanding The Behavior Of 5, 10, 15, 20-Tetrakis (4 -Hydroxyphenyl) Porphyrin and Its Cation in Methanol: Insights from Electronic Structure Calculations. J.Con.Ma. 2023;1(2):163-165
. Available From https://jcm.thecmrs.in/index.php/j/article/view/27 |
 |
 ©
©